TopFIND
TopFIND web interface
https://topfind.clip.msl.ubc.ca
TopFIND is the Termini oriented protein Function Inferred Database (TopFIND) is an integrated knowledgebase focused on protein termini, their formation by proteases and functional implications. It contains information about the processing and the processing state of proteins and functional implications thereof derived from research literature, contributions by the scientific community and biological databases. TopFIND has been conceived and developed by Philipp Lange during his postdoctoral research in the Overall Lab at UBC.
Background: Among the most fundamental characteristics of a protein are the N- and C-termini defining the start and end of the polypeptide chain. Genetically encoded termini can vary due to alternative splicing and novel protein termini isoforms are also by alternative translation. Once proteins are translated, termini are highly dynamic, being frequently trimmed at their ends by a large array of exopeptidases. Neo-termini can also be generated by endopeptidases after precise and limited proteolysis, termed processing. Necessary for the maturation of many proteins, processing can also occur afterwards, often resulting in dramatic functional consequences. Aberrant proteolysis can cause wide range of diseases like arthritis or cancer. Hence, proteolytic generation of pleiotrophic stable forms of proteins, the universal susceptibility of proteins to proteolysis, and its irreversibility, distinguishes proteolysis from many highly studied posttranslational modifications.
Kowledgebase content: TopFIND is a resource for comprehensive coverage of protein N- and C-termini discovered by all available in silico, in vitro as well as in vivo methodologies. It makes use of existing knowledge by seamless integration of data from UniProt, MEROPS (protease cleavage), Ensembl (alternative splicing) and TisDB (alternative translation) and provides access to new data from community submission and manual literature curating. It renders modifications of protein termini, such as acetylation and citrulination, easily accessible and searchable and provides the means to identify and analyse extend and distribution of terminal modifications across a protein.
Data access: The data can be accessed through protein specific pages with a strong emphasis on the relation to curated background information and underlying evidence that led to the observation of a terminus, its modification or proteolytic cleavage. In brief the protein information, its domain structure, protein termini, terminus modifications and proteolytic processing of and by other proteins is listed. All information is accompanied by metadata like its original source, method of identification, confidence measurement or related publication. A positional cross correlation evaluation matches termini and cleavage sites with protein features (such as amino acid variants) and domains to highlight potential effects and dependencies in a unique way. Also, a network view of all proteins showing their functional dependency as protease, substrate or protease inhibitor tied in with protein interactions is provided for the easy evaluation of network wide effects. A powerful yet user friendly filtering mechanism allows the presented data to be filtered based on parameters like methodology used, in vivo relevance, confidence or data source (e.g. limited to a single laboratory or publication). This provides means to assess physiological relevant data and to deduce functional information and hypotheses relevant to the bench scientist. The data can also be accessed for lists of termini or protease substrates using the software tools TopFINDer and PathFINDer:
TopFINDer and PathFINDer
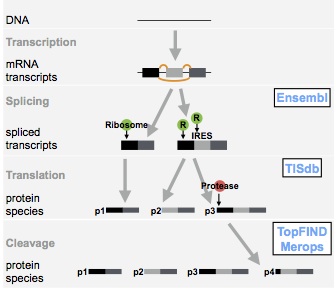 The TopFIND ExploreR, TopFINDer (link): TopFINDer allows for the access of TopFIND data for large lists of proteomics-derived N- or C-termini. It automatically characterizes and annotates these lists for their sequence context and position, genesis (cleavage, alternative splicing, or alternative translation), and implications for protein structure and function. Additionally to retrieving these data for each terminus in the list, TopFINDer further reports protease statistics relevant to the list for the identification of dominant proteases in the sample. Read more in the publication.
The TopFIND ExploreR, TopFINDer (link): TopFINDer allows for the access of TopFIND data for large lists of proteomics-derived N- or C-termini. It automatically characterizes and annotates these lists for their sequence context and position, genesis (cleavage, alternative splicing, or alternative translation), and implications for protein structure and function. Additionally to retrieving these data for each terminus in the list, TopFINDer further reports protease statistics relevant to the list for the identification of dominant proteases in the sample. Read more in the publication.
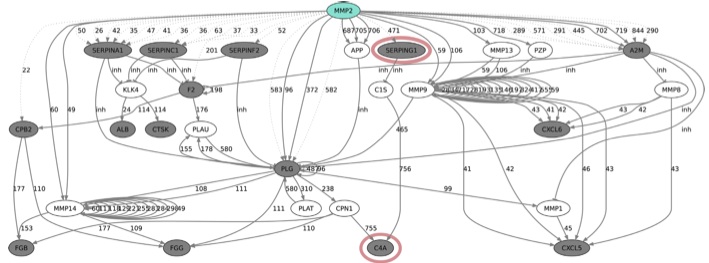 PathFINDer (link): PathFINDer identifies indirect connections between a protease and list of substrates or termini thus supporting the evaluation of complex proteolytic processes in vivo. The protease web is a network of proteases and their inhibitors. Proteases cleave other proteases and inhibitors of other proteases in vivo. Changes of the activity of one protease can thereby influence the cleavage of substrates of other proteases indirectly. PathFINDer uses a graph to model known protease interactions as a network and finds paths in this network. These paths represent biological pathways thus enabling mechanistic insights. Read more in the publication.
PathFINDer (link): PathFINDer identifies indirect connections between a protease and list of substrates or termini thus supporting the evaluation of complex proteolytic processes in vivo. The protease web is a network of proteases and their inhibitors. Proteases cleave other proteases and inhibitors of other proteases in vivo. Changes of the activity of one protease can thereby influence the cleavage of substrates of other proteases indirectly. PathFINDer uses a graph to model known protease interactions as a network and finds paths in this network. These paths represent biological pathways thus enabling mechanistic insights. Read more in the publication.
The development of TopFINDer and PathFINDER by N. Fortelny were led by Philipp Lange during his postdoctoral research in the Overall Lab at UBC.